
For decades, developing treatments for rare diseases, which are medical conditions that affect fewer than 200K people at any given time, were viewed as one of the least attractive markets for pharmaceutical development. Patient populations were too small, development costs too high, and commercial upside too limited to justify investment. The Orphan Drug Act of 1983 changed that by restructuring the economics of drug development through tax credits, market exclusivity, and accelerated regulatory pathways, turning rare disease from a neglected corner of medicine into a viable commercial category.
Since then, however, the rare disease market has evolved into a commercial and scientific priority for pharmaceutical developers beyond that which policymakers originally envisioned. Rare disease is no longer just a niche area of biotech supported only by government incentives; it has become the primary testing ground for new therapeutic platforms such as gene editing, RNA interference, antisense oligonucleotides, and gene therapies. Small, genetically defined patient populations allow companies to validate entirely new treatment modalities faster and with greater biological precision before expanding those technologies into the larger markets of more common diseases.
As a result, the central priorities in developing rare disease treatment are shifting. The principal challenge is no longer simply determining whether a company can build a successful “orphan drug”, which is a pharmaceutical agent developed specifically to treat a rare medical condition. Instead, the question is whether a company can use rare disease to validate a scalable platform technology, extend that platform into broader indications, and navigate the reimbursement dynamics created by multimillion-dollar curative therapies.
Orphan drugs are a critical contributor to this shift towards platform strategies more broadly, and research on orphan drugs has already benefited drug development in general. The companies best positioned for the next decade will not be those treating orphan drugs as isolated products, but those using rare disease as the commercial and regulatory proving ground for the next generation of biotech platforms.
The History of Orphan Drugs in the US
The term "orphan disease" is an economic designation rather than a clinical one. These conditions were orphaned not necessarily by providers or researchers, but by pharmaceutical companies hesitant to invest hundreds of millions of dollars over a decade in drugs that would serve small populations consisting of thousands of patients. Orphan drug development requires $1–2 billion or more to bring a single treatment to market, and recouping that investment requires either a large patient population or higher prices than insurers and government programs want to pay.
Somewhere between 7-10K rare diseases collectively affect 30 million Americans, more than cancer and heart disease combined. In the early 1980s, when the Orphan Drug Act was passed, 95% of these diseases had no FDA-approved treatment. In lieu of a commercial incentive to create drugs for these diseases, policies have been proposed and passed to nationally fund and develop such drugs.
Government agencies, however, have historically been ill-equipped for this role. The FDA regulates medicines, but doesn't directly or indirectly incentivize their creation. The NIH funds basic research, but it isn’t designed to run large-scale clinical trials or manage commercial-scale manufacturing. Direct government development of drugs would also eliminate the competitive pressure that drives innovation, replacing market efficiency with bureaucratic process.
The 1983 Orphan Drug Act (ODA) took a different approach. Rather than developing orphan drugs itself, the government introduced new incentives for the development and manufacturing of drugs for rare diseases. It created a formal designation process to recognize drugs targeting rare diseases, which they defined as those affecting fewer than 200K people in the United States (or any condition for which there is no reasonable expectation of commercial profitability for other reasons). It also attached concrete financial incentives to that designation, including tax credits for clinical testing, waivers of multi-million-dollar FDA user fees, and seven years of market exclusivity upon approval.
How the Orphan Drugs Act Reset Incentives
Tax credits for the costs of qualified clinical trials for orphan-designated drugs were originally set at 50%, and later reduced to 25% by the 2017 Tax Cuts and Jobs Act. Unlike a deduction, which reduces taxable income, a tax credit directly reduces the taxes a company owes, making it especially valuable for smaller biotech firms with limited capital. Since clinical trials are the most expensive phase of drug development, this credit effectively transfers a portion of the downside risk of a trial's failure to the government.
Orphan-designated drugs are also exempt from FDA user fees. The FDA charges several million dollars in application fees when companies submit a drug for approval. Removing these fees meaningfully reduces upfront costs that could otherwise deter smaller companies or those pursuing multiple rare disease indications simultaneously.
Once an orphan drug is approved, a seven-year window begins during which the FDA will not approve a competing drug for the same disease. This creates a temporary, legally protected monopoly, giving the sponsor the pricing power necessary to recover its R&D investment without immediate competitive pressure. This window is designed to be long enough to attract private investment but short enough to eventually allow competition and market correction.
Beyond financial incentives, the ODA also introduced expedited review pathways that shorten the time from development to patients. Fast Track allows more frequent FDA interaction during development. Breakthrough Therapy provides intensive FDA guidance for drugs showing early clinical promise. Priority Review cuts the standard 12-month review clock to six months. None of these programs changes the evidentiary standard for approval.
The Long-Term Impact of the ODA
The next few decades showed that the ODA was successful in incentivizing orphan drug development. By August 2018, 503 unique medicines targeting 731 orphan indications had been approved, and 78% of them were approved solely for orphan diseases. By 2023, more than 370 orphan drugs were actively marketed (some approved drugs were later discontinued, withdrawn for safety reasons, or never commercially launched).
Not only that, but the pace accelerated over time: between 1983 and 2019, over 5K drugs and biologics received orphan drug designation, with the number of designations more than doubling in the 1980s and 1990s, nearly doubling between the 1990s and 2000s, and nearly tripling between the 2000s and 2010s.
The effects of orphan drug development went beyond orphan diseases. Research into some rare diseases led to new, often more efficient drug development processes. About 80% of rare diseases are monogenic (caused by a single genetic mutation), making the underlying biology easier to characterize and target. Genetically defined patient populations can also be easier to diagnose via genetic testing, allowing companies to run smaller, more efficient trials with cleaner endpoints and faster paths to regulatory validation.
The diseases that attracted the most development efforts in the decades since the passage of the ODA share a common profile: a well-characterized genetic mechanism, clear biomarkers or measurable clinical endpoints, a patient population large enough to support trials, and an engaged patient community advocating with regulators. Together, these conditions made rare disease not just more commercially viable under the ODA's incentive structure, but more scientifically productive per dollar invested.
The Drug Development Payer Problem
The reimbursement system for drugs is built around the basic assumption that drugs are taken repeatedly, so costs are recurring and can be spread over time. Despite the incentives introduced by ODA, this reimbursement model makes it economically challenging to break even for some rare disease treatments.
Small molecules and RNA interference (RNAi) both fit that description because they modify existing biology, correcting the impact of a faulty protein or silencing a problematic gene, but the underlying genetic defect remains, and treatment continues indefinitely. Genetic treatments, however, work differently, correcting or replacing the gene itself in a single intervention.
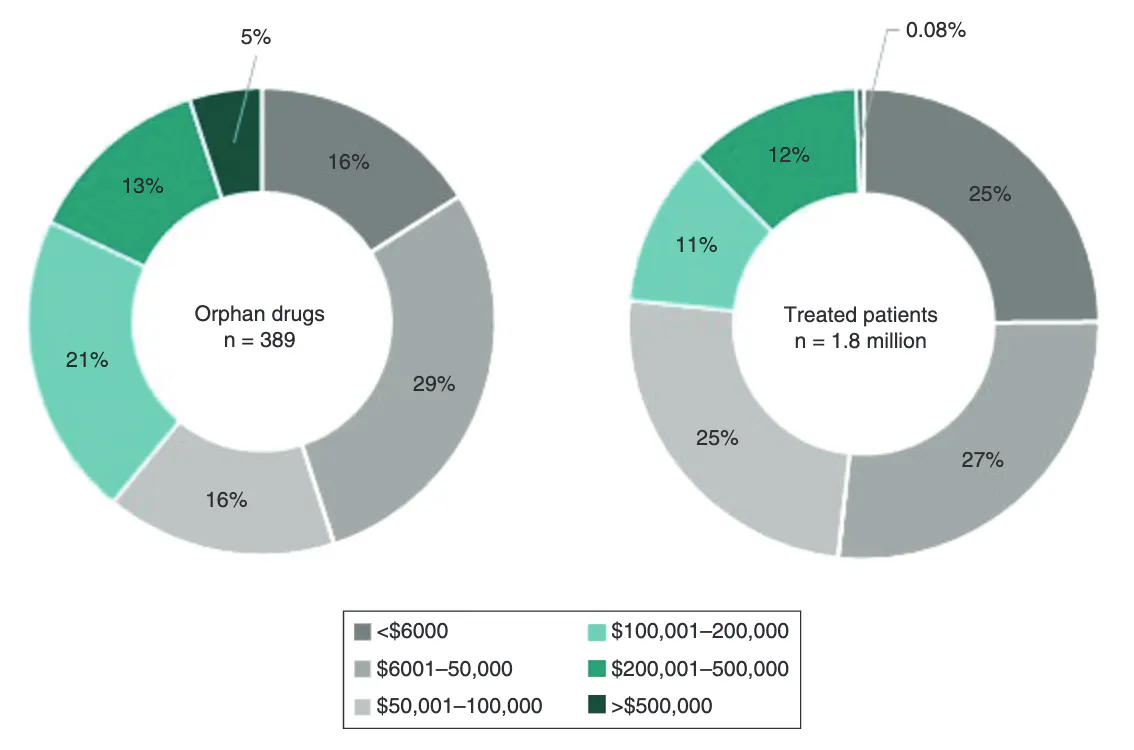
For this reason, many orphan drugs carry high list prices: 39% cost more than $100K annually, and gene and cell therapies in particular routinely run into the hundreds of thousands of dollars or more. In 2019, the average annual cost of an orphan treatment per treated patient was $32K, though individual treatments ranged anywhere from $6K to $500K per year.
The trajectory of these costs has been steep rather than incremental. The US saw a 26-fold increase in total orphan drug costs between 1998 and 2017, with annual cost growth running at roughly 12% since.
The economic structure of insurers, annual premiums collected against annual costs, makes these costs difficult to absorb. A drug that costs $3 million once but saves $500K a year in avoided ongoing care can be cost-effective over a patient's lifetime, but the insurer paying that $3 million today may not be the same insurer collecting the savings five years later, once the patient has changed jobs or insurance plans. This mismatch means no individual payer has a strong incentive to pay full price for a cure, even when the cure is, in aggregate, a good deal for the healthcare system. This is a problem for any expensive one-time therapy, not just those specific to rare diseases.
Platform Drug Development
This mismatch has pushed the drug development industry towards a platform strategy for getting new technologies to market in the first place. The biotech industry is undergoing a structural shift from single-asset companies, which develop one drug for one disease, to platform companies, which build a reusable technology that can be applied to many diseases in succession. A platform, in this context, is not a drug, but the underlying engine that discovers and produces drugs. Platform companies recycle the same core technology, applied repeatedly, to produce a stream of new drugs rather than just one.
Platform companies benefit from compounding returns: each successful drug makes the next one cheaper and faster to develop, because the core technology, manufacturing process, and regulatory relationship with the FDA are already established. This lowers drug development costs, leading to lower drug costs for patients and insurers. Examples of drug development platforms include a chemistry and screening process that can be re-run against new disease targets, a delivery mechanism that can carry different genetic payloads to the same tissue, or a manufacturing and vector system that can package different genes for gene therapy.
Rare disease has become the preferred arena to prove that these platforms work, for a specific scientific reason: roughly 80% of rare diseases are caused by a single-gene mutation, so the drug target is known from the outset. Many of these diseases have well-established biomarker endpoints, such as protein levels, enzyme activity, and lung function, that can demonstrate a platform is working biologically long before a company has the years of follow-up needed to show clinical outcomes.
Biotechnology companies such as Vertex and Alnylam each demonstrate this model, but in two different layers of drug development: one validates a drug discovery platform, the other a drug delivery platform.
Case Study: Vertex Pharmaceuticals
Vertex is a pharmaceutical company that developed a drug discovery platform during the development of its cystic fibrosis (CF) drug, Trifakta, which was used by 90% of all CF patients by November 2023.
CF affects roughly 100K people worldwide and is caused by mutations in the CFTR gene, which produces a misfolded protein that disrupts chloride transport and clogs the lungs with thick mucus. As of 2006, conventional wisdom held that the CFTR protein itself was too difficult to target directly, so treatments were confined to managing symptoms rather than halting disease progression.
CFTR mutations fall into distinct functional classes: some prevent the channel from opening properly (gating defects), others prevent the protein from folding and reaching the cell surface at all (trafficking defects), and others reduce the channel's conductivity once it's there. Vertex built a screening system using robotic assays capable of testing hundreds of thousands of chemical compounds for their ability to restore the essential chloride transport in CFTR-deficient cells, paired with a chemistry process to take promising "hits" from that screen and optimize them into viable drugs. This combination forms a repeatable process that could be re-run against each new mutation class to produce a new compound.
The first product from the Vertex platform was ivacaftor (Kalydeco), approved in 2012. It targeted the G551D mutation, a gating defect, by restoring the channel's function rather than just treating symptoms. This defect affects about 4% of CF patients, or roughly 1.2K people in the US. Because the underlying research engine worked, Vertex re-ran it against other mutation classes, producing a sequence of drugs rather than a single one.
Orkambi in 2015 and Symdeko in 2018 added "correctors," compounds that fix protein folding and trafficking, to address the more common F508del mutation, which causes a different type of defect than G551D. Trikafta (2019), a three-drug combination, extended coverage to roughly 90% of all CF patients. Each new drug came from the screening-and-chemistry process applied to a different class of mutations, not from modifying a single drug. As of July 2026, Vertex had expanded into adjacent technologies, including gene editing and other next-generation modalities.
Case Study: Alnylam Pharmaceuticals
Whereas Vertex demonstrates the platform thesis in drug discovery, Alnylam demonstrates it in a different layer of the business: drug delivery. RNA interference (RNAi) works upstream of where small molecules operate, silencing defective genes before problematic proteins are made.
In the human body, every protein is produced from instructions carried by a messenger RNA (mRNA) molecule, which is itself copied from a gene. RNAi drugs are short synthetic RNA fragments (siRNA) engineered to intercept and destroy the messenger before the cell can manufacture the protein.
In principle, this means any disease-causing gene could be silenced. These drugs have a broader scope than small molecules, which depend on a protein having a structural pocket for them to bind to. The practical obstacle for such drugs is the fragility of RNA, which degrades in the bloodstream within seconds and cannot cross cell membranes unassisted. Alnylam's platform addresses this delivery obstacle by attaching siRNA molecules to GalNAc (N-acetylgalactosamine), a sugar that binds with high affinity to the asialoglycoprotein receptor (ASGPR) protein.
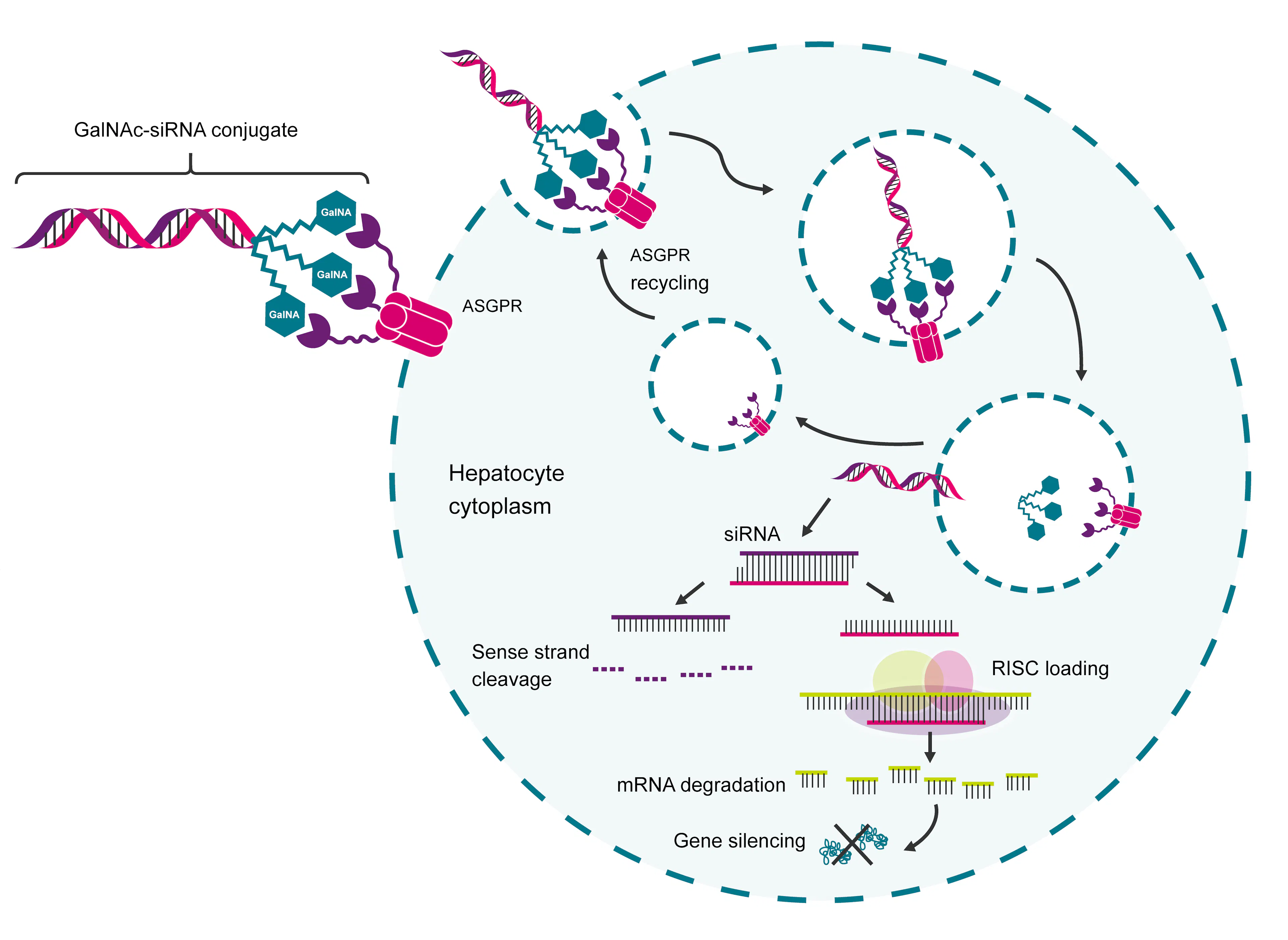
Source: Biosearch Technologies
This protein is expressed almost exclusively, and in very high numbers, on liver cells. Once a GalNAc-siRNA conjugate binds ASGPR, the liver cell engulfs it through a process called endocytosis; the receptor recycles back to the cell surface to repeat the process, while the siRNA payload is released inside the cell to begin silencing its target gene. This GalNAc "chassis" is the platform that enables transport of any RNAi drug into the liver.
The first validation of this delivery platform was for hATTR amyloidosis, a disease caused by a misfolded protein produced almost entirely by the liver. This disease affects roughly 50K people worldwide, is uniformly progressive, and had no disease-modifying treatment prior to Alnylam’s development, which was approved in August 2018.
Alnylam next used the platform for treatment of diseases driven by PCSK9, a liver-produced protein that raises LDL cholesterol and is implicated in cardiovascular disease. The resulting drug, Leqvio, was approved in the US in 2021 and uses the same GalNAc-ASGPR delivery mechanism with a different siRNA payload.
Case Study: Sarepta Pharmaceuticals
Duchenne Muscular Dystrophy (DMD) is an X-linked genetic disease affecting roughly 10 in 100K people. It's caused by mutations in the dystrophin gene, which prevents production of dystrophin, a protein that keeps muscle cells structurally intact. Without it, muscle tissue progressively breaks down.
Sarepta's first approach to DMD was exon skipping. The dystrophin gene is made up of 79 exons, the coding segments that get stitched together to produce the final protein. In most DMD patients, one or more exons are missing, which interferes with the reading frame and results in a nonfunctional protein. Exon-skipping drugs use short synthetic genetic sequences called antisense oligonucleotides (ASOs) to make the cell's protein-building machinery skip over the damaged section, restoring the reading frame and producing a shorter, partially functional dystrophin protein. This approach is mutation-specific and less generalizable: each exon requires its own distinct drug. Sarepta built separate ASOs for exons 51, 53, and 45, but those three drugs together cover only about 30% of DMD patients.
Sarepta's second approach broke with that pattern by adopting a more broadly applicable drug-as-a-platform for the dystrophin gene. Elevidys, approved in 2023, is a gene therapy that delivers a shortened but functional copy of the dystrophin gene directly into muscle cells using an adeno-associated virus (AAV) as the delivery vector. Unlike exon skipping, Elevidys is mutation-agnostic: because it supplies a working gene rather than patching the existing broken one, it works for the vast majority of DMD patients regardless of which specific exon is affected, with only narrow exceptions. It also requires just one infusion rather than a lifetime of repeated dosing.
AAV-based gene therapy still doesn't correct the underlying mutation; it adds a functional copy of the gene alongside the broken original, rather than fixing the original itself. That creates two practical problems. The added gene's expression can fade over time, since it isn't permanently woven into the patient's own DNA the way a true correction would be. And because the immune system mounts a response to the AAV vector, patients who receive one AAV-based gene therapy generally cannot safely receive a second — there's effectively one shot at this style of treatment per patient. These limitations are the direct motivation for the next generation of gene-editing tools, which aim to fix the mutation itself rather than add a workaround alongside it.
Elevidys epitomizes both the platform thesis and the challenges that rare disease drug developers face. The treatment costs $3.2 million per patient; a one-time treatment at that price doesn't fit into a reimbursement system built around recurring, modest annual costs, making it hard for insurers to cover. Sarepta is left with a therapy that may be scientifically sound but is not yet commercially viable, meaning the company will likely have to extend its drugs’ applications to other genes to take advantage of the platform thesis financially.
It is also worth noting that Elevidys’s accelerated approval was controversial because of a lack of evidence that it actually worked, and multiple patient deaths after approval, which were attributed to acute liver failure. In July 2025, the FDA placed Sarepta's investigational gene therapy trials on clinical hold following these three deaths, revoked the company's platform technology designation, and requested it voluntarily stop all shipments of Elevidys.
The Future of Orphan Drug Development
Vertex, Alnylam, and Sarepta trace a progression in how platform therapies can be developed and evolve, from platform drug discovery to drug delivery to broadly-applicable individual drugs like AAV. As of June 2026, the rare drug industry is increasingly considering solutions that address the limitations of these platforms.
Base editing, developed by David Liu at Harvard (for which he won the 2025 Breakthrough Prize), is the logical next step because it solves the durability and one-shot problems that AAV delivery still faces. Standard CRISPR-based editing cuts both strands of the DNA double helix, and that double-strand break creates room for errors during the cell's repair process, including unintended deletions or rearrangements.
Base editing avoids that risk entirely: rather than cutting the DNA, it chemically converts one base directly into another at a specific location in the genome. Because the change is made directly to the patient's own DNA rather than delivered alongside it, it doesn't fade over time the way an AAV-delivered gene can, and it doesn't carry the same one-time-use constraint. That precision makes it especially well-suited to diseases caused by a single point mutation, rather than a missing exon or a broken protein; roughly 30% of known disease-causing mutations are the kind base editing can directly correct.
Prime editing, also developed by Liu, extends that same logic further. Where base editing can only convert one base to another, prime editing can perform insertions, deletions, and all twelve possible base-to-base conversions. Prime editing remains earlier in clinical development than base editing, but it represents the same underlying trajectory of each generation of technology, moving the intervention one step closer to the original genetic error.
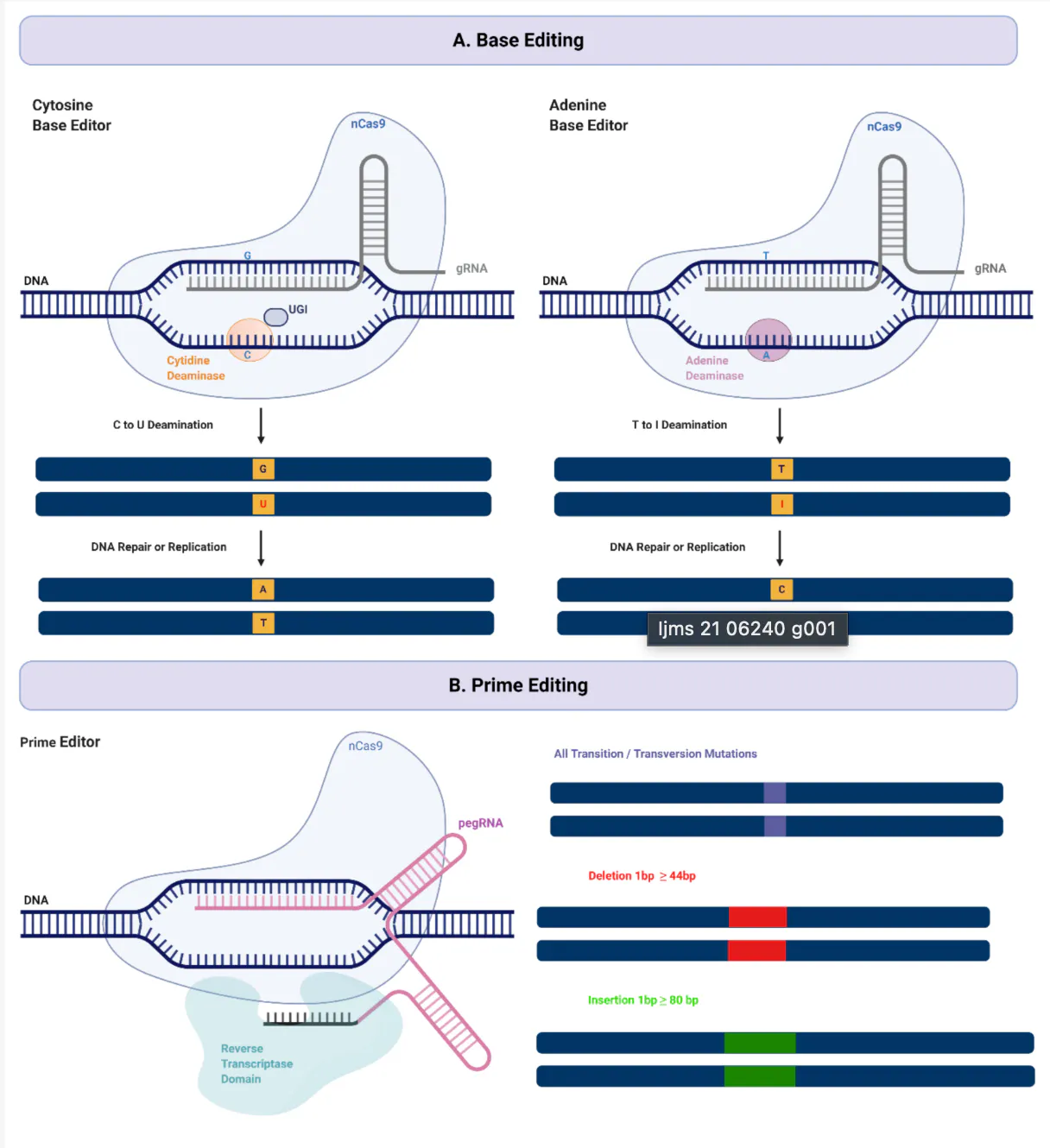
Source: MDPI
This same progression is also relevant for the payer problem. The mismatch described earlier exists because a one-time cure and an annual-premium insurance system are structurally incompatible. While editing technologies don't resolve that mismatch on their own, a correction made directly to a patient's genome, with no added viral vector and no risk of fading expression, is a more durable claim of "cured" than what AAV-based gene therapy can currently promise, making insurers more likely to underwrite one-time seven-figure prices.
Capital has increasingly shifted toward platform biotech broadly, spanning protein engineering and AI-driven drug discovery alongside gene editing and RNA therapeutics, precisely because investors have drawn the same conclusion: a platform that works once can be made to work again, faster and cheaper each time, while a single-asset drug cannot.
The Orphan Drug Act made the development of treatments for rare diseases commercially viable in 1983, and generations of platforms have since used it to develop drugs that would otherwise never have existed. What remains unresolved is whether reimbursement can evolve to match the delivery mechanisms for these treatments. The companies that define the next decade of rare disease will be the ones that can address the financing gap in addition to the biology itself by building platforms that compound with each approval and using the biology of a rare disease as a beachhead into a larger one.

